Spectral methods for clustering protein sequences
 a weighted
undirected graph in which each node corresponds to a protein sequence
and the weights on the edges correspond to a measure of distance
between two sequences. The goal is to partition such a graph into a set
of discrete clusters whose members are homologs. Formulating the
problem in this way it was possible to analyze the limitations that one
might expect from earlier methods and I was able to provide a
theoretical explanation of why spectral clustering methods should
perform better for this problem. Then, I introduced a spectral method
that partitions the graph into clusters by considering the random walk
formulation on the graph, and analyzing the perturbations to the
stationary distribution of a Markov
a weighted
undirected graph in which each node corresponds to a protein sequence
and the weights on the edges correspond to a measure of distance
between two sequences. The goal is to partition such a graph into a set
of discrete clusters whose members are homologs. Formulating the
problem in this way it was possible to analyze the limitations that one
might expect from earlier methods and I was able to provide a
theoretical explanation of why spectral clustering methods should
perform better for this problem. Then, I introduced a spectral method
that partitions the graph into clusters by considering the random walk
formulation on the graph, and analyzing the perturbations to the
stationary distribution of a Markov 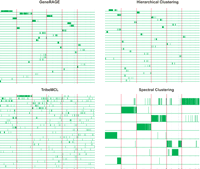 relaxation process. This is done by
looking
at the eigenvectors of the Markov transition matrix. The weights on the
edges of the graphs that I used are a nonlinear transformation of the
pairwise BLAST e-values, and the parameters of such nonlinear
transformation were learned. I tested this algorithm on difficult sets
of proteins whose relationships are known from the SCOP database [1, 3,
4]. My method correctly identified many of the family/superfamily
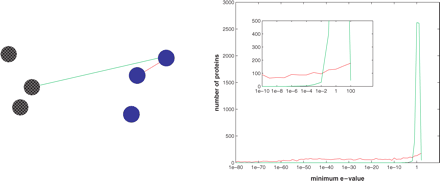
relationships. I found that the eigenvalue spectrum and in particular,
the eigengaps provide useful indication on the number of clusters in
the dataset. Using this approach the results obtained were much better
than those obtained using other methods on the same datasets. In fact,
I consistently observed that, the number of clusters obtained for a
given set of proteins was close to the number of superfamilies in that
set; there were fewer singletons; and the method correctly grouped most
remote homologs. In my experiments, the quality of the clusters as
quantified by a measure that combines sensitivity and specificity was
consistently better (on average, improvements were: 84% over
Hierarchical Clustering, 34% over Connected Component Analysis (similar
to GeneRAGE), and 72% over TribeMCL. In
collaboration
with Rajkumar Sashidaran (Mark
Gerstein's lab,
relaxation process. This is done by
looking
at the eigenvectors of the Markov transition matrix. The weights on the
edges of the graphs that I used are a nonlinear transformation of the
pairwise BLAST e-values, and the parameters of such nonlinear
transformation were learned. I tested this algorithm on difficult sets
of proteins whose relationships are known from the SCOP database [1, 3,
4]. My method correctly identified many of the family/superfamily
relationships. I found that the eigenvalue spectrum and in particular,
the eigengaps provide useful indication on the number of clusters in
the dataset. Using this approach the results obtained were much better
than those obtained using other methods on the same datasets. In fact,
I consistently observed that, the number of clusters obtained for a
given set of proteins was close to the number of superfamilies in that
set; there were fewer singletons; and the method correctly grouped most
remote homologs. In my experiments, the quality of the clusters as
quantified by a measure that combines sensitivity and specificity was
consistently better (on average, improvements were: 84% over
Hierarchical Clustering, 34% over Connected Component Analysis (similar
to GeneRAGE), and 72% over TribeMCL. In
collaboration
with Rajkumar Sashidaran (Mark
Gerstein's lab, [1] A. Paccanaro, J. A. Casbon, M. A. S. Saqi (2006). Spectral Clustering of Proteins Sequences Nucleic Acids Research 2006 Mar 17;34(5):1571-80 [pubmed]
[2] R
Sasidharan, M Gerstein, A Paccanaro
(2006) Spectral Clustering of Protein
Sequences Using Sequence-Profile Scores Proceeding of ICNPSC 2006 —
third
International Conference on Neural Parallel and Scientific
Computations,
Atlanta, USA [pdf]
[4] C.
Chennubhotla, A. Paccanaro
(2003). Markov Analysis of Protein
Sequence Similarities Lecture Notes in Computer Science series,
Vol. 2859,
278-286, Springer-Verlag. 14 [pdf]
Techniques for integrating different protein-protein interaction experiments
 mainly other proteins. Thus, protein
interactions provide an
important clue to the function of proteins. Many interaction datasets,
mostly
from large-scale experiments are available now. However, the quality of
protein
interaction experiments varies greatly with the researcher who performs
the
experiment and with the particular technique used. Therefore, it is
important
to be able to integrate the results of different experiments into a
more
reliable measurement. Together with Haiyuan Yu, I developed a
system that provided such
reliable
measurement by combining the results of different experiments through a
set of
learned weights. Using the hand-curated protein complexes in
the MIPS reference database we trained a system to
assign a probability that each pairwise interaction is true based on
experimental reproducibility and mass spectrometry scores from the
relevant purifications. Thus from the raw experimental data we obtained
a protein–protein interaction network as an undirected weighted graph
in which individual proteins are nodes and the weight of the edge
connecting two nodes is the probability that the interaction is
correct. I am currently attempting to improve these results by
including into the measurement information about the network topology.
[This
work is done in collaboration with the laboratory of Andrew
Emili's lab,
mainly other proteins. Thus, protein
interactions provide an
important clue to the function of proteins. Many interaction datasets,
mostly
from large-scale experiments are available now. However, the quality of
protein
interaction experiments varies greatly with the researcher who performs
the
experiment and with the particular technique used. Therefore, it is
important
to be able to integrate the results of different experiments into a
more
reliable measurement. Together with Haiyuan Yu, I developed a
system that provided such
reliable
measurement by combining the results of different experiments through a
set of
learned weights. Using the hand-curated protein complexes in
the MIPS reference database we trained a system to
assign a probability that each pairwise interaction is true based on
experimental reproducibility and mass spectrometry scores from the
relevant purifications. Thus from the raw experimental data we obtained
a protein–protein interaction network as an undirected weighted graph
in which individual proteins are nodes and the weight of the edge
connecting two nodes is the probability that the interaction is
correct. I am currently attempting to improve these results by
including into the measurement information about the network topology.
[This
work is done in collaboration with the laboratory of Andrew
Emili's lab, Krogan et al, (2006), Global landscape of protein complexes in the yeast Saccharomyces cerevisiae Nature, 2006, Mar 30,440(7084):637-43 [pubmed]
Methods for denoising large scale protein-protein interaction experiments
Large-scale experiments for the identification of Protein-Protein interactions are known to be error-prone. I have developed two methods for inferring protein-protein interactions which are
 based purely on topological properties of
interaction networks observed experimentally and can correct many of
the errors present in large-scale experiments. The basic idea of the
first method (joint work with Haiyuan
Yu, Harvard University and Valery Trionov, Yale
University) derives from the way in which large-scale experiments are
carried out and particularly from the matrix model interpretation of
their results. In these experiments, one protein (the bait), is used to
pull out the set of proteins interacting with it (the preys), i.e. its
protein complex, in the form of a list. When such lists differ only in
a few elements, it is reasonable to assume that this is because of
experimental errors, and the missing elements should therefore be
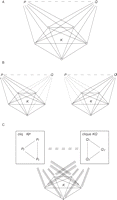
added. Each list can be represented as a fully connected graph in which
proteins occupy the nodes. Then the problem of identifying lists that
differ in only a few elements is equivalent to finding a clique with a
few missing edges, which we called a "defective clique". Therefore the
algorithm searches the network for defective cliques (i.e. nearly
complete complexes of pairwise interacting proteins) and predicts the
interactions that complete them. The second method (which I am
currently refining), computes a
diffusion
distance between each pair of proteins and then infers an interaction
when such
distance is below a given threshold. Both methods have been shown to
have a very good predictive performance, thus allowing the correction
of many errors present in large-scale experiments.
based purely on topological properties of
interaction networks observed experimentally and can correct many of
the errors present in large-scale experiments. The basic idea of the
first method (joint work with Haiyuan
Yu, Harvard University and Valery Trionov, Yale
University) derives from the way in which large-scale experiments are
carried out and particularly from the matrix model interpretation of
their results. In these experiments, one protein (the bait), is used to
pull out the set of proteins interacting with it (the preys), i.e. its
protein complex, in the form of a list. When such lists differ only in
a few elements, it is reasonable to assume that this is because of
experimental errors, and the missing elements should therefore be
added. Each list can be represented as a fully connected graph in which
proteins occupy the nodes. Then the problem of identifying lists that
differ in only a few elements is equivalent to finding a clique with a
few missing edges, which we called a "defective clique". Therefore the
algorithm searches the network for defective cliques (i.e. nearly
complete complexes of pairwise interacting proteins) and predicts the
interactions that complete them. The second method (which I am
currently refining), computes a
diffusion
distance between each pair of proteins and then infers an interaction
when such
distance is below a given threshold. Both methods have been shown to
have a very good predictive performance, thus allowing the correction
of many errors present in large-scale experiments.H. Yu, A.
Paccanaro, V. Trifonov, M. Gerstein (2006). Predicting
interactions
in
protein networks
by completing defective cliques. Bioinformatics 2006 Apr
1;22(7):823-9 [pubmed]
A.
Paccanaro, V. Trifonov, H. Yu, M. Gerstein (2005). Inferring
Protein-Protein Interactions Using
Interaction Network Topologies. — IJCNN 2005 - International Joint
Conference on Neural Networks,
Prediction of protein-protein interactions
L. J. Lu,
Y. Xia, A. Paccanaro, H. Yu, M. Gerstein
(2005). Assessing the Limits of
Genomic Data Integration for Protein-Protein Interactions Genome
Research,
Jul 2005; 15: 945 – 953 [pubmed]
Prediction of gene essentiality from genomic features
Predicting essential genes remains a main goal in directed drug design for antimicrobial or antifungal targets. Currently, most essential gene prediction is performed by homology searches to organisms where essentiality is known and has been experimentally tested. While quick and easy, this system is also simplistic. Together with Michael Seringhaus (Mark
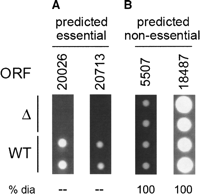 Gerstein's Lab, Yale University), we aimed
at
improving the efficacy of such prediction through the integration of
genomic-scale data, and the application of machine learning techniques.
We trained
a classification system on S. cerevisiae, where
the Saccharomyces Genome Deletion project has ascertained essentiality
for 95%+
of the genome. For each gene in the organism, we collected a set of
genomic
features – some derived from sequence information, others from
functional
genomics experiments. We used these data to learn a system that
can
predict essential genes in S. cerevisiae.
We then applied this system to three recently-sequenced yeast genomes (S. bayanus, S. mikatae, and C. albicans)
for which essential genes
have not been experimentally identified. We then compared our
predictive engine
to a simple BLAST homology search, and a subset of our putative
essential
candidates in S. bayanus and S. mikatae
were tested with knockouts in vivo. We were able to
demonstrate for the first time that it is possible to learn traits
associated
with essential genes in yeast species and to use these features in a
predictive
manner. Our approach therefore shows promise for the identification of
drug
targets in novel and pathogenic species. We are currently continuing
this work
by studying the relative importance and effects of the different types
of features
on the prediction. [This work is done in collaboration with the
laboratories of Michael Snyder
and Mark Gerstein,
Gerstein's Lab, Yale University), we aimed
at
improving the efficacy of such prediction through the integration of
genomic-scale data, and the application of machine learning techniques.
We trained
a classification system on S. cerevisiae, where
the Saccharomyces Genome Deletion project has ascertained essentiality
for 95%+
of the genome. For each gene in the organism, we collected a set of
genomic
features – some derived from sequence information, others from
functional
genomics experiments. We used these data to learn a system that
can
predict essential genes in S. cerevisiae.
We then applied this system to three recently-sequenced yeast genomes (S. bayanus, S. mikatae, and C. albicans)
for which essential genes
have not been experimentally identified. We then compared our
predictive engine
to a simple BLAST homology search, and a subset of our putative
essential
candidates in S. bayanus and S. mikatae
were tested with knockouts in vivo. We were able to
demonstrate for the first time that it is possible to learn traits
associated
with essential genes in yeast species and to use these features in a
predictive
manner. Our approach therefore shows promise for the identification of
drug
targets in novel and pathogenic species. We are currently continuing
this work
by studying the relative importance and effects of the different types
of features
on the prediction. [This work is done in collaboration with the
laboratories of Michael Snyder
and Mark Gerstein, M Seringhaus, A Paccanaro, A Borneman, M Snyder and M Gerstein (2006) Predicting Essential Genes in Fungal Genomes, Genome Research, 2006, 16 (9): 1126-35 [pubmed]
Predicting protein function in E. Coli
 We performed
an extensive proteomic survey using affinity-tagged E. coli strains and
generated comprehensive genomic context inferences to derive a
high-confidence compendium for virtually the entire proteome. We used
an N-step diffusion method to assign un-annotated genes to specific
biological processes.
We performed
an extensive proteomic survey using affinity-tagged E. coli strains and
generated comprehensive genomic context inferences to derive a
high-confidence compendium for virtually the entire proteome. We used
an N-step diffusion method to assign un-annotated genes to specific
biological processes.[This work is done in collaboration with the laboratory of Andrew Emili,
P. Hu, S. C. Janga, M. Babu, J. J. D’iaz-Mej’ia, G. Butland, W. Yang, O. Pogoutse, X. Guo, S. Phanse, P. Wong, S. Chandran, C. Christopoulos, A. Nazarians-Armavil, N. K. Nasseri, G. Musso, M. Ali, N. Nazemof, V. Eroukova, A. Golshani, A. Paccanaro, J. F. Greenblatt, G. Moreno-Hagelsieb, and A. Emili Global functional atlas of Escherichia coli encompassing previously uncharacterized proteins PLoS biology, vol. 7, iss. 4, p. 1000096, 2009].
[pubmed]
[this paper was highlighted in Nature Methods 2009; 6, 402-403]
Quantifying environmental adaptation of metabolic pathways in metagenomics
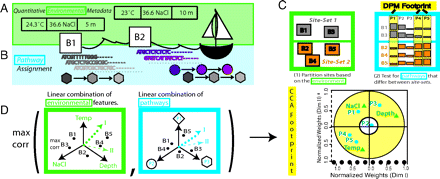 Recently, approaches have been developed
to sample the genetic content of heterogeneous environments
(metagenomics).
Recently, approaches have been developed
to sample the genetic content of heterogeneous environments
(metagenomics).We investigated how these sequences link distinct environmental conditions with specific biological processes.
We focused on how the usage of particular pathways and subnetworks reflects the adaptation of microbial
communities across environments and habitats. We introduced a novel approach to relate multiple, continuously
varying factors defining an environment to the extent of particular microbial pathways present in a geographic site.
Furthermore, we developed a method to identify ensembles of weighted pathways which we called metabolic footprints
that are predictive of their environment and could be used as biosensors.
[This work is done in collaboration with the laboratories of Mark Gerstein and Michael Snyder,
T. A. Gianoulis, J. Raes, P. V. Patel, R. Bjornson, J. O. Korbel, I. Letunic, T. Yamada, A. Paccanaro, L. J. Jensen, M. Snyder, P. Bork, and M. B. Gerstein Quantifying environmental adaptation of metabolic pathways in metagenomics, Proceedings of the National Academy of Sciences, vol. 106, iss. 5, pp. 1374-1379, 2009]. [pubmed]
[this paper was highlighted in Science 2009; 323 (5918) and Nature Genetics 2009; 41 (275)]